

???? Introduction: Why It Matters
Complaint handling and CAPA (Corrective and Preventive Action) systems are a regulatory safety net within medical device firms.
Strong systems convert customer complaints, field failures, audits, and nonconformances into structured improvements that protect patients, motivate product quality, and result in FDA compliance.
Without robust systems:
❌ FDA Form 483 observations and warning letters can close down your company.
❌ Frequent product failure can cause recalls and lawsuits.
❌ Poor quality can destroy your reputation and investors’ trust.
A lean system that is compliant ensures:
✅ Regulatory trust.
✅ Patient safety.
✅ Ongoing improvement.
✅ Scalability of your MedTech company.
???? Complaint handling refers to
FDA 21 CFR 820.198 states that a complaint is:
Any verbal, electronic, or written report that reports defects in the device’s identity, quality, durability, reliability, safety, effectiveness, or performance after it has been made available for distribution.
Significance:
Legally required for post-marketing surveillance of safety and effectiveness.
It facilitates the early identification of field issues, thereby preventing their escalation.
This system enables Medical Device Reporting (MDR) for reportable adverse events.
Examples:
A physician reports a loose wearable sensor while in use.
A patient contacts us by email concerning an error code on a product being used at home.
A distributor reports multiple returns with the same fault.
In doubt, document and evaluate.
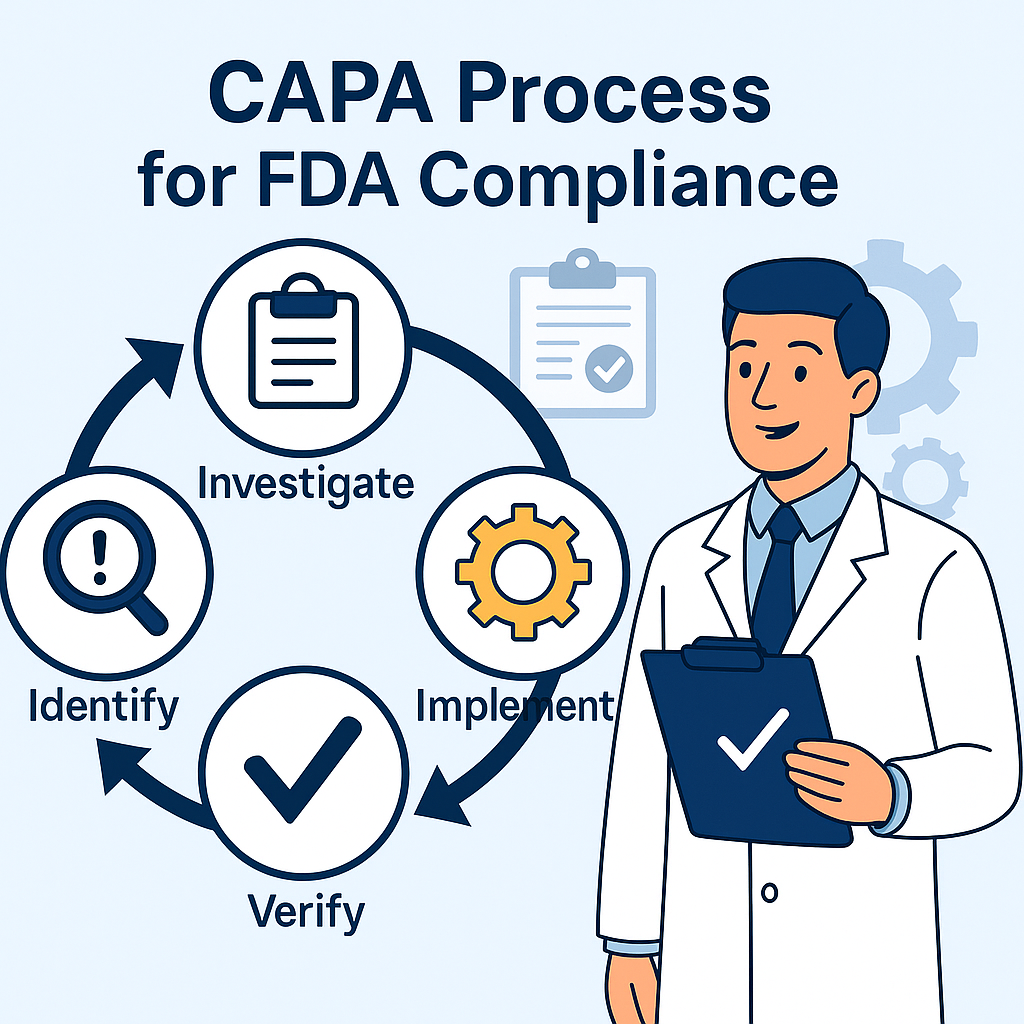
???? What is CAPA?
CAPA (Corrective and Preventive Action), by 21 CFR 820.100, is the official process for:
✅ Corrective Action: Correcting known quality issues to avoid recurrence.
✅ Preventive Action: Identifying and removing potential problems before they occur.
Why it is essential:
FDA considers CAPA a pillar of your QMS.
It drives systemic correction, no bandages.
Resolving the root causes of CAPAs minimizes the cost of poor quality.
Examples:
A series of complaints of battery failure triggers a CAPA to look into, correct, and prevent reoccurrence.
A series of low-level nonconformances on inspections triggers preventative process changes.
???? How to Set Up an Acceptable Complaint Handling System
1️⃣ Define and Document
Create a complaint-handling SOP with a complaint definition.
Train all to recognize complaints (calls, emails, verbal complaints) and escalate promptly.
2️⃣ Intake and Logging
Use a Complaint Intake Form or eQMS system recording:
Complainant details.
The details of the device, such as its model and lot/serial number, are crucial.
Record the date, time, and method of receiving the complaint.
The nature of the complaint varies.
Record complaints immediately upon their receipt.
3️⃣ Triage for MDR
Verify if the complaint is MDR-reportable under 21 CFR Part 803:
Did it result in or lead to death/serious injury?
Would failure most likely cause serious injury/death if it happened again?
4️⃣ Investigate
The process involved retrieving as many devices as possible, obtaining them, and reviewing them.
Examine production history records and DHR (Device History Record).
The interview involved staff as required.
Use formal investigation aids (5 Whys, Fishbone).
5️⃣ Take Corrective Actions if Required
If you identify a systemic cause, link the complaint with a CAPA.
Record all actions taken because of the specific complaint.
6️⃣ Communicate and Close
Notify the complainant of closure as applicable.
Obtain QA sign-off before closure.
Maintain archive records for FDA inspection readiness.
7️⃣ Trend and Analyze
Review complaint data routinely to identify repeat issues.
Use data to launch CAPAs and improve design/manufacturing processes.
???? How to Develop an Effective CAPA System
1️⃣ Identify Data Sources
Capture possible issues from:
Complaints.
Nonconformances (NCRs).
Audit findings.
Supplier issues.
Field/service reports.
2️⃣ Prioritize Issues
Use risk-based thinking:
High-risk and high-frequency problems will require immediate implementation of CAPA.
3️⃣ Investigate Root Causes
Gather objective evidence.
Utilize structured tools (5 Whys, Fishbone, Fault Tree Analysis).
Document extensively for traceability.
4️⃣ Develop and Implement Actions
For Corrective Actions:
Revise SOPs.
Retrain personnel.
Modify manufacturing processes.
Design changes.
For Preventive Actions:
Add additional process checks or monitoring.
Improve supplier controls.
Design changes.
5️⃣ Verify Effectiveness
Collect post-implementation information.
Monitor applicable KPIs to verify resolution.
Watch for unintended effects.
6️⃣ Document and Close
Your CAPA file must include the following:
Problem description.
Investigation details.
The root cause analysis is conducted.
We have implemented corrective and preventive actions.
Verification/validation of effectiveness.
QA and management must approve the closure.
????️ FDA Expectations During Inspections
FDA will review:
✅ Investigation timeliness and complaint history.
✅ Compliance with MDR reporting.
✅ Ensure that CAPAs include thorough root cause analysis and proper closure procedures.
The process involves managing risks and incorporating design updates.
FDA would like to see these systems as a closed-loop quality improvement process.
???? Frequent Blunders to Avoid
❌ Not recording or ignoring oral complaints.
❌ Weak investigations without facts.
❌ Delayed MDR reporting.
❌ CAPAs are never to be closed.
❌ Closing CAPAs without confirmation.
❌ Inadequate documentation, with no way of verifying compliance.
???? Best Practices for Long-Term Compliance
✅ Well-defined SOPs with tasks and deadlines.
✅ Educate employees to recognize and forward complaints.
✅ Use structured RCA regularly.
✅ Ensure effectiveness before closing CAPA.
✅ Regularly check trend data during management reviews.
✅ Maintain clean, inspection-ready records.
???? Real-World Example
A new business producing wearable devices had recurring Bluetooth connection complaints. Initially:
❌ Addressed issues reactively by replacing the device.
No trend or investigation was conducted. ❌
At the time of FDA inspection:
The company received a Form 483 because it failed to handle complaints adequately and did not have any CAPAs in place.
The firm:
✅ Implemented formal recording of complaints and SOPs.
✅ Analyzed the data using the 5 Whys method, which helped identify firmware defects and issues with supplier tolerances.
✅ Implemented a firmware fix and supplier corrective actions.
✅ Verified their effectiveness using post-market surveillance.
Outcome: Complaint levels were reduced by 85%, and the firm avoided a warning letter.
Advantages of Strong Complaint and CAPA Systems
✅ Maintain patient safety and confidence.
✅ Reduce quality issues and return to the field.
✅ Prevent costly recalls and liability.
✅ Be prepared for FDA inspection.
✅ Drive continuous product and process improvement.
📚 References
1️⃣ FDA 21 CFR Part 820 – Quality System Regulation
🔗 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820
2️⃣ FDA 21 CFR 820.198 – Complaint Files Regulation
🔗 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/subpart-M/section-820.198
3️⃣ FDA 21 CFR 820.100 – Corrective and Preventive Action Regulation
🔗 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/subpart-J/section-820.100
4️⃣ FDA 21 CFR Part 803 – Medical Device Reporting (MDR)
🔗 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-803
5️⃣ FDA QSIT (Quality System Inspection Technique) Guide
🔗 https://www.fda.gov/media/80177/download
6️⃣ ISO 13485:2016 – Medical Devices Quality Management Systems (Purchase or preview)
🔗 https://www.iso.org/standard/59752.html
7️⃣ FDA Device Advice – Postmarket Requirements (Complaints & MDR)
🔗 https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/postmarket-requirements-devices
8️⃣ FDA Guidance: Quality System Information for Certain Premarket Application Reviews
🔗 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/quality-system-information-certain-premarket-application-reviews
📢 Coming Next:
“UDI Compliance for Medical Devices: A Step-by-Step Implementation Guide.”
You will learn:
✅ FDA UDI requirements and practical implementation.
✅ Labeling, data submission, and system integration.
✅ Avoiding UDI mistakes that delay launch and scaling.